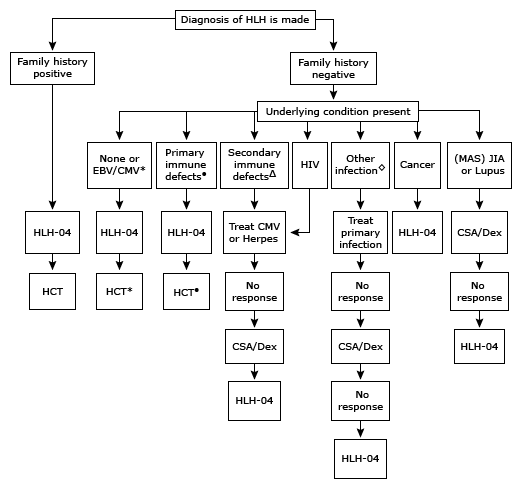
Note: HLH-2004 is a research treatment protocol. The HLH-94 trial, described in the text, should be used in those being treated outside of a therapeutic research trial.
HLH: hemophagocytic lymphohistiocytosis; MAS: macrophage activation syndrome; EBV: Epstein-Barr virus; CMV: cytomegalovirus; HIV: human immunodeficiency virus; JIA: juvenile idiopathic arthritis; HLH-04: HLH 2004 treatment protocol; CSA/Dex: treatment with cyclosporin + dexamethasone; XLP: X-linked lymphoproliferative syndrome; HCT: hematopoietic cell transplantation.
* Check for mutations listed below. Since EBV, CMV, herpes, parvovirus, and many other viral infections can unmask HLH caused by one of these mutations, do NOT use presence of these infections to delay treatment with HLH-04. HCT is recommended for all patients <2 years of age, those with CNS-HLH, poor response to the first 8 weeks of HLH-04 (or relapse), and all those with homozygous mutations of perforin, MUNC13-4, XLP, and syntaxin genes.
• Griscelli & Chediak-Higashi syndromes, XLP, Kawasaki disease. Rashes of HLH may resemble Kawasaki-type rashes. HCT is needed for the first three of these diseases and for Kawasaki disease if poor response to HLH-04.
Δ Solid organ or bone marrow transplantation.
◊ Bacterial or fungal infection, leishmaniasis, malaria.
HLH-04 protocol
PubMed
TIHLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis.
AUHenter JI, Horne A, AricóM, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G
SOPediatr Blood Cancer. 2007;48(2):124.
In HLH-94, the first prospective international treatment study for hemophagocytic lymphohistiocytosis (HLH), diagnosis was based on five criteria (fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, and hemophagocytosis). In HLH-2004 three additional criteria are introduced; low/absent NK-cell-activity, hyperferritinemia, and high-soluble interleukin-2-receptor levels. Altogether five of these eight criteria must be fulfilled, unless family history or molecular diagnosis is consistent with HLH. HLH-2004 chemo-immunotherapy includes etoposide, dexamethasone, cyclosporine A upfront and, in selected patients, intrathecal therapy with methotrexate and corticosteroids. Subsequent hematopoietic stem cell transplantation (HSCT) is recommended for patients with familial disease or molecular diagnosis, and patients with severe and persistent, or reactivated, disease. In order to hopefully further improve diagnosis, therapy and biological understanding, participation in HLH studies is encouraged.
ADChildhood Cancer Research Unit, Department of Woman and Child Health, Karolinska University Hospital, Karolinska Institutet, Stockholm, Sweden. Jan-Inge.Henter@ki.se
HCT
TI Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis.
AUHorne A, Janka G, Maarten Egeler R, Gadner H, Imashuku S, Ladisch S, Locatelli F, Montgomery SM, Webb D, Winiarski J, Filipovich AH, Henter JI, Histiocyte Society
SOBr J Haematol. 2005;129(5):622.
Haemophagocytic lymphohistiocytosis (HLH) poses major therapeutic challenges, and the primary inherited form, familial haemophagocytic lymphohistiocytosis (FHL), is usually fatal. We evaluated, including Cox regression analysis, survival in 86 children (29 familial) that received HLH-94-therapy (etoposide, dexamethasone, ciclosporin) followed by allogeneic stem cell transplantation (SCT) between 1995 and 2000. The overall estimated 3-year-survival post-SCT was 64% [confidence interval (CI) = +/-10%](n = 86); 71 +/- 18% in those patients with a matched related donor (MRD, n = 24), 70 +/- 16% with a matched unrelated donor (MUD, n = 33), 50 +/- 24% with a family haploidentical donor (haploidentical, n = 16), and 54 +/- 27% with a mismatched unrelated donor (MMUD, n = 13). After adjustment for potential confounding factors, estimated odds ratios (OR) for mortality were 1.93 (CI =0.61-6.19) for MUD, 3.31 (1.02-10.76) for haploidentical, and 3.01 (0.91-9.97) for MMUD, compared with MRD. In children with active disease after 2-months of therapy (n = 43) the OR was 2.75 (1.26-5.99), compared with inactive disease (n = 43). In children with active disease at SCT (n = 37), the OR was 1.80 (0.80-4.06) compared with inactive disease (n = 49), after adjustment for disease activity at 2-months. Mortality was predominantly transplant-related. Most HLH patients survived SCT using MRD or MUD, and survival with partially mismatched donors was also acceptable. Patients that responded well to initial pretransplant-induction therapy fared best, but some persisting HLH activity should not automatically preclude performing SCT.
ADChildhood Cancer Research Unit, Institution for Woman and Child Health, Karolinska Institutet, Department of Paediatric Haematology and Oncology, Karolinska Hospital, Stockholm, Sweden.